Getting the app for your operating system & installation¶
Getting the app¶
Prebuilt app¶
Most users (Windows, Mac, Linux) should download the prebuilt jar file (executable app) available at
HpoCaseAnnotator releases.
To run HpoCaseAnnotator, you need to have Java Runtime Environment (Java version 8) installed on the machine.
To run the app, a double click should work. Alternatively, enter following at the command line.
$ java -jar HpoCaseAnnotator.jar
Build from sources¶
It is also possible to build the HpoCaseAnnotator from source code using Maven:
$ git clone https://github.com/monarch-initiative/HpoCaseAnnotator.git
$ cd HpoCaseAnnotator
$ mvn package
After successful build process the HpoCaseAnnotator.jar will be present in hpo-case-annotator-gui/target directory.
Initial setup¶
After a successful startup the dialog window will be opened:
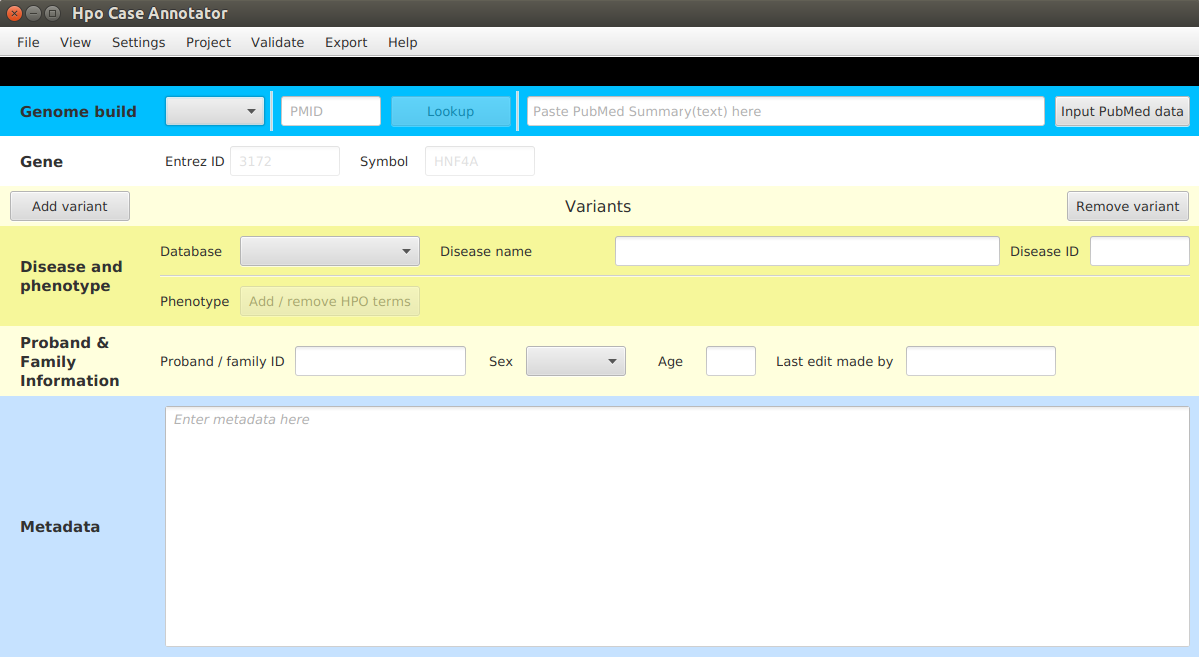
Note, that not all the functionality is enabled after the first startup, since there are some resources that need to
be downloaded first. Click on Settings | Set resources to start setting up the app.
A new dialog window will be opened:
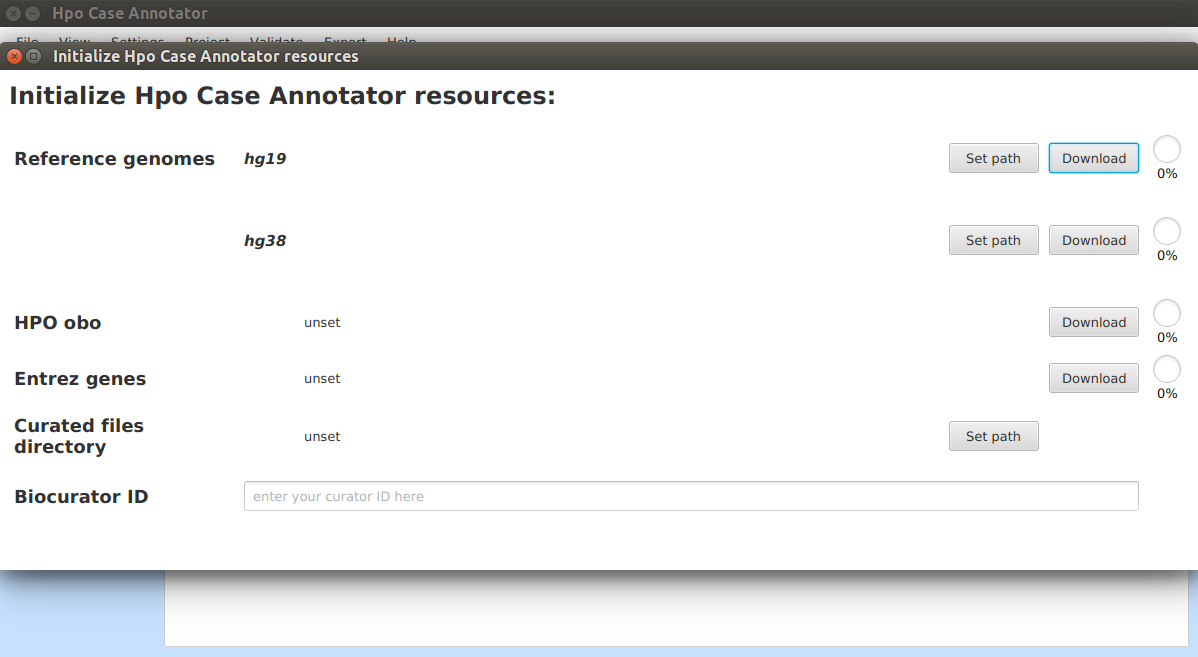
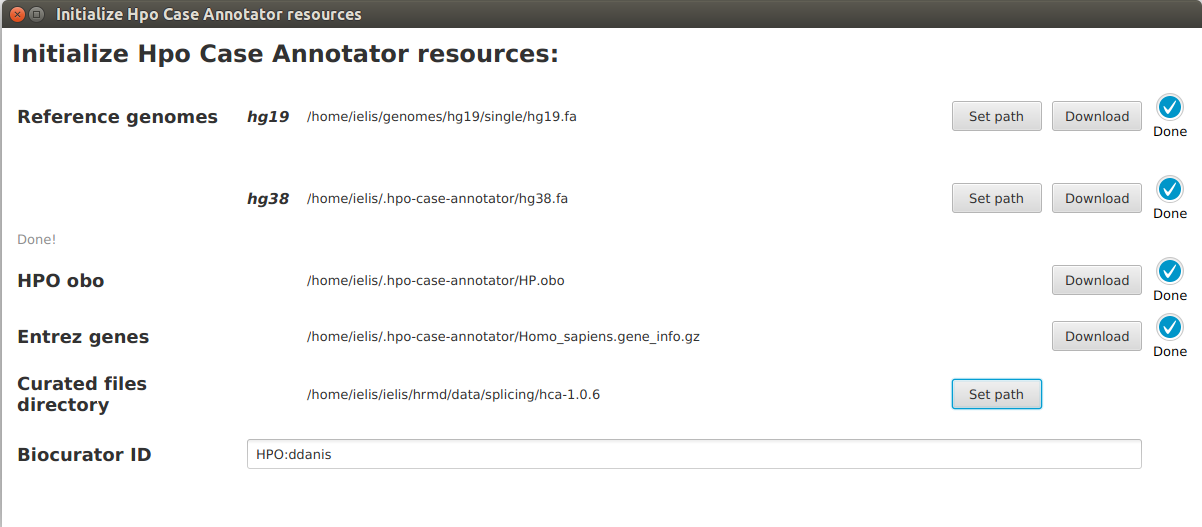
Reference genome¶
Hpo Case Annotator needs access to the sequence of the reference genome in order to e.g. check whether the wildtype sequence entered for each variant matches the corresponding genomic position. Hpo Case Annotator is able to download and pre-process the reference genome or you can provide the FASTA file yourself.
Currently, GRCh37 (hg19) and GRCh38 (hg38) genome assemblies are supported. For our internal use, we are still at hg19, and will use a liftover to move to hg38 coordinates!
- Download and pre-process the reference genome sequences automatically
In order to download the reference genome click on the Download buttons of individual genome assemblies. Each assembly file has roughly 1 GB and the download process may take up to 20 minutes depending on the speed of your internet connection. Do not close before the download & pre-processing is completed. After successful download, sequences of all the chromosomes will be concatenated into a single FASTA file and index will be created automatically using HTS-JDK library.
- Provide FASTA file
You do not have to download the reference genome files if there are already some present in your system. Use Set path buttons to set paths to local FASTA files. Corresponding index (*.fai) files should be present in the same directory.
This is all we need for the genomic position Q/C routines.
HPO obo¶
The app will automatically download the newest version of Human Phenotype Ontology (HPO) in OBO format, the file has ~5 MB. This needs to be done once (and can be updated as necessary). We download a copy of the OBO file so that the App can autocomplete the HPO terms names and labels.
Entrez genes¶
The app will also download information regarding genes. This is also a one-time operation and after the download autocompletion of gene symbols and IDs will be available.
Curated files directory¶
Each curated case is stored as a file in JSON format. Here we set path to a directory where the JSON files created in a single project are stored.
Biocurator ID¶
Here provide your biocurator ID.
All of the resources are required to be downloaded just once and after these steps the app is fully prepared for work.